Creating controlled release drugs, which release medicine at a specific rate or location in the digestive system, can be difficult. Physiologically-based pharmacokinetic (PBPK) modeling is a useful tool for this. It considers the drug’s physical and chemical properties and existing data on how the drug acts in the body. This helps predict how the drug will dissolve and behave in the body, guiding the development of the drug formula.
This extensive summary explains the advantages of controlled release drugs, how to choose drugs for this method, the role of PBPK modeling in developing these drugs, and creating modified release products for the best patient results.
WHAT ARE CONTROLLED RELEASE DOSAGE FORMS?
Controlled release medications are designed to slowly release their active ingredients, ensuring a steady effect and less frequent doses. These drugs, often taken orally, control how fast the active ingredient is released, limiting how quickly it’s absorbed. By targeting specific areas in the digestive tract, the drug’s behavior in the body can be tailored to each project’s needs. Such formulations keep drug levels in the blood between the minimum effective and maximum tolerated concentrations, offering several benefits over immediate release version including:
- Prolonged effectiveness
- Increased drug half-life
- Improved efficacy
- Reduced dosing frequency
- Reduced side effects.
IN VIVO PREDICTIVE DISSOLUTION TOOLS TO ESTABLISH IN VITRO–IN VIVO CORRELATIONS
In vitro-in vivo correlation (IVIVC) is defined as a mathematical model that describes the relationship between an in vitro property of a dosage form (i.e., dissolution) and its in vivo response (i.e., exposure). The utility of an IVIVC depends on how accurately in vivo exposure can be predicted based on in vitro data. An effective IVIVC is dependent on the design of the in vitro and in vivo studies that are used to develop and validate it.
The in vitro dissolution data is collected using one of several dissolution apparatuses which can vary, based on the dissolution media and set up. Dissolution media can range from simple buffered solutions to those containing bile salts, lipid digestion products and/or enzymes. Compendial dissolution setups such as a basket or a paddle have the advantage of being simple, robust, and used worldwide.
Traditional methods may not fully mimic the changing conditions a drug faces in the digestive system, like varying pH and fluid composition. To address this, researchers have developed advanced techniques like the gastrointestinal simulator (GIS). The GIS, which has been effective in studying drugs like posaconazole, uses separate chambers to simulate different parts of the digestive system and adds fluids at specific rates to replicate changing conditions. This setup, combined with computer models using known drug data, can predict how different drug formulations will perform in the body. This helps in selecting the most promising drug prototypes for clinical trials.
wHICH DRUG IS A SUITABLE CANDIDATE FOR CONTROLLED RELEASE?
Several factors determine whether a drug is suitable for controlled release dosage forms, including:
- Dose
- Dose to solubility ratio
- Stability
- Absorption mechanism- passive diffusion preferred
- Regional permeability- colonic absorption important.
- PK half life
- Metabolism.
Suitability for modified release formulations and prototype performance can be determined using an appropriate IVIVC; however, often there is a lack of data required to establish an IVIVC. In such instances, PBPK modelling is a highly useful alternative to further evaluate the feasibility of developing a successful controlled release formulation.
PBPK models can be developed using the following approaches:
- Top-down approach: Models built predominantly on the observed clinical data.
- Bottom-up approach: Models based on knowledge of the Absorption, Distribution, Metabolism and Excretion (ADME) processes within the human body along with the physicochemical and in vitro data of an API.
- Middle-out approach: Combining bottom-up and top-down approaches to build the model and validate it against the available clinical data.

The middle-out approach is typically preferable, as it provides a balance between the top-down and bottom-up approaches while leveraging enough data to be accurate.
Often, the middle-out approach will entail parameter optimization using data from animal PBPK models or PBPK models for similar drugs, providing a good prediction of the performance.
How can PBPK models be used to determine suitability for controlled release?
- Step 1: Conduct a physicochemical evaluation and developability assessment of the drug.
- Step 2: Proceed with PBPK modelling, using the middle-out approach to build a model that fits the available in vivo data. Use the model to verify absorption in the colon, as this is a key factor determining in vivo performance of controlled release formulations.
- Step 3: Based on the desired therapeutic range, predict the in vitro dissolution profile that would give the desired exposure, and provide it to the formulation team as the target in vitro dissolution release profile for development work.
DEVELOPMENT OF MODIFIED RELEASE PRODUCTS
Modified release drug products are intentionally designed to alter the rate or timing of drug release and include controlled release, extended release, delayed release and others. When developing a ‘modified release dosage form’, a good scientific approach consisting of a thorough understanding of the API physiochemical properties as well as the excipient properties is imperative in designing a robust product. In addition, it is also important to have a general understanding of the target patient population, i.e.:
- Pediatric, adult or geriatric
- Does the patient population have disease specific challenges that can affect compliance
In general, to develop a product development strategy for a controlled release dosage form, most developers adopt a stepwise approach through the employment of four well defined steps:
1. Scientific assessment: The assessment of API physicochemical characteristics, namely, dose, solubility, particle size, stability and potential for absorption in the lower gastrointestinal tract.
2.Target product profile (TPP): To ensure alignment across key stakeholders, it is important to have a well-planned TPP that defines the critical attributes of the drug product as well as describing how it will be suitable for use by the target patient population. This will not only help accelerate development timelines but will also minimize development risks, and eventually lead to an optimal product for the patient.
3.Development concept: Once the TPP is established, the product development team can start building on a conceptual design of the formulation component and manufacturing process of the dosage form.
4.Controlled release prototypes: Once suitable prototypes are identified, dissolution tests can be conducted to compare performance and elucidate drug release mechanisms. Preclinical testing of lead prototypes in suitable in vivo models could help increase the chance of identifying formulations that will work in the clinic.
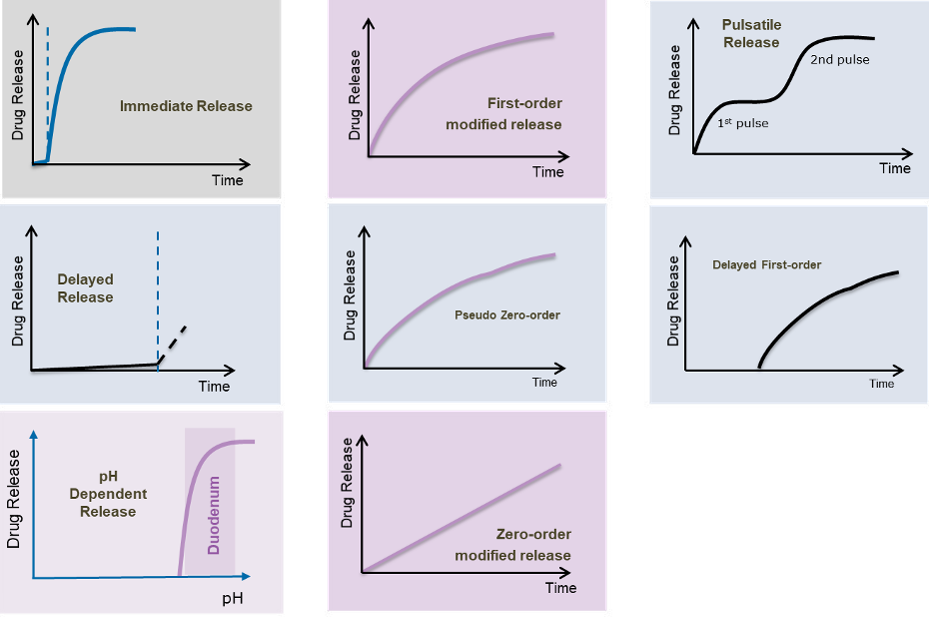
ACHIEVING DIFFERENT TYPES OF MODIFIED RELEASE PROFILES
Beyond the basic ‘immediate release’ profile, various modified release profiles are achievable with different colors. These include
- First order modified release
- Delayed first order
- Pulsatile release
- pH dependent release
- Delayed release •
- Zero-order modified release.
- Pseudo zero-order
Depending on the intended site of drug absorption in the gastrointestinal tract and the excipients used, the drug can be formulated in a single unit or multi-particulate dosage form. Matrix or coated type systems can be used in both of these dosage forms to achieve different types of drug release profiles. The dose and the solubility of the API determine whether a coated or a matrix development approach is most appropriate for a given program, i.e.:
- A matrix system better suits low solubility and high dose APIs
- A coated system better suits high solubility and high dose APIs.
There are pros and cons for each technology which need to be considered when developing dosage units. A single unit tablet, either coated or matrix type, is simple to produce and well suited for commercial manufacturing. However, a single unit tablet has a less flexible drug release mechanism.
In addition, failure of a single unit system may cause rapid release of the drug (i.e., dose dumping) which can be a major safety risk.
On the other hand, multi-particulate dosage forms, such as sugar cores layered with drug and functional polymer, matrix pellets produced via wet mass extrusion/ spheronization or mini tablets, could mitigate the risk of dose dumping because the failure of a few units will have less impact on the patient. These multi-particulates can provide flexible dosing options and reproducible release profiles as they are typically less dependent on gastric emptying.
In conclusion, the importance of understanding the API’s physiochemical properties and the selection of the right manufacturing technology can help in designing different dosage forms with different release profiles for improved patient outcomes.

HOW RENEJIX CAN HELP?
By matching each project to a knowledgeable and experienced team, Hycon Pharma ensures all work is performed according to an agreed timeline and budget.
As experts in PBPK modelling, Hycon Pharma scientists can help speed up the development of dosage forms with release profiles that deliver the desired PK performance. During the development of modified release dosage forms, Hycon Pharma product development teams consider the patients’ needs, the TPP and the properties of the drug substance to ensure selection of the formulation strategy best suited for the program.
Whether seeking controlled-release technology to extend a product’s lifecycle, or because a specific pharmacokinetic profile is needed, Hycon Pharma can help achieve your project goals.
For more information visit: www.renejix.com





