“New drugs receiving expedited approval have shorter timelines, which can create unique challenges for development and manufacturing.
Joe Dukich, director of operations at Rene Pharma shares insights on manufacturing strategies for these expedited programs where there might be just one chance to get it right.There are four designations by the U.S. Food and Drug Administration (FDA) to provide momentum for drug development and approval: Fast Track, Breakthrough Therapy.
Accelerated Approval, and Priority Review. Per the FDA, each approach intends to expedite the availability of drugs that might treat serious conditions, especially drugs that are the first of their kind or that significantly improve upon existing treatments.
Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fill an unmet medical need. The purpose is to get important new drugs to the patient earlier. Fast Track addresses a broad range of serious conditions. Breakthrough Therapy designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s).
A Typical Drug Product Development Life Cycle
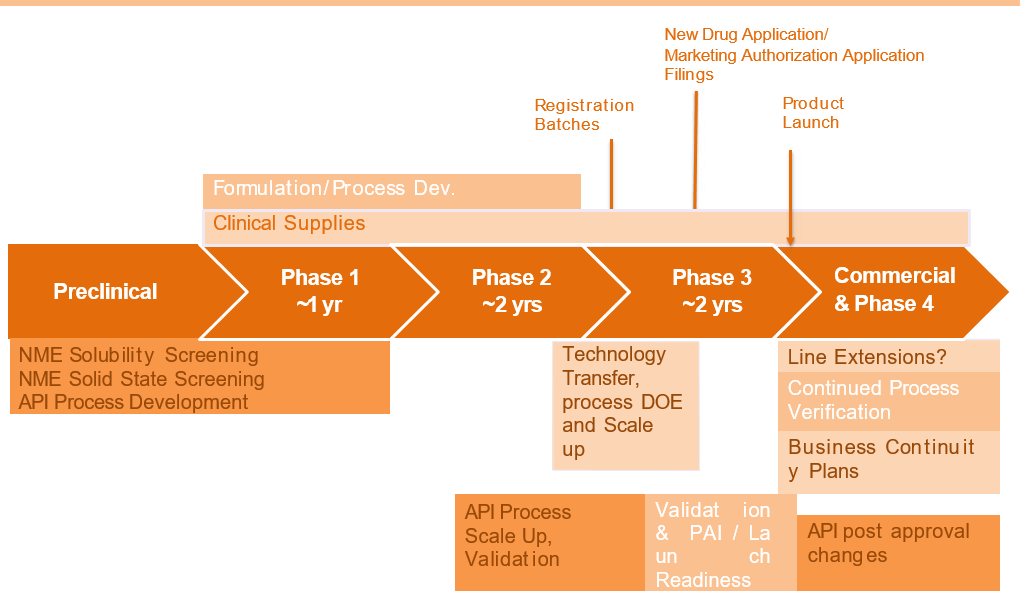
Accelerated Approval designation allows drugs for serious conditions that fill an unmet need to be approved based on a surrogate endpoint and thus enables the FDA to approve these drugs faster. A Priority Review designation means the FDA’s goal is to act on an application within 6 months (compared to 10 months under standard review).
Whatever the program’s designation, there is a rise in the overall number of expedited development programs to address patients’ needs, so how is our industry adapting to the drug development life cycles? Drug development life cycles for these designations involve interwoven steps and related processes be executed within an accelerated timeline. The challenge is to leverage the drug development knowledge and build a strategy that maintains the integrity, quality, and timeliness of the manufacturing process.
Here we will highlight the key strategies involved in overcoming manufacturing challenges associated with expedited development programs.
How To Manage Stability Programs To Support Accelerated Development Timelines?
Stability testing provides the much-needed insight on how formulation and manufacturing processes can affect the API over time. To keep up with the accelerated development timelines, it is important to define the given drug substance’s attributes clearly to understand how it might affect the quality and stability of the final drug product. For instance, insights derived from solid state characterization and polymorph screening can be incorporated in the formulation process design. This will help avoid setbacks that can result if manufacturing is unaware of any potential polymorphic changes. Likewise, it is important to design a formulation process to avoid potential degradation pathways or disruptions by selecting compatible excipients and setting the appropriate excipient and API specifications (i.e., any potential issues during manufacturing and packaging, such as photosensitivity, moisture, and/or temperature variability, and the possibility of oxidation).
By taking a strategic approach to dose form selection and considering the relative advantages of simple versus complex formulations in development can save time and cost in an accelerated development pathway. By selecting a simple dosage form initially and planning simultaneously can assist in moving the drug quickly to the post-approval stage. For example, if Phase 1 stability data is available for capsules, it can be leveraged for approval. An alternative is using the preliminary information to explore the option of dosing in a tablet form, which will grant the development team time to generate adequate stability data.
The impact of packaging, shipping and storage conditions should also be studied to determine any potential effects on the stability.
FIT-FOR-PURPOSE MANUFACTURING STRATEGIES TO SUPPORT AN ACCELERATED TIMELINE
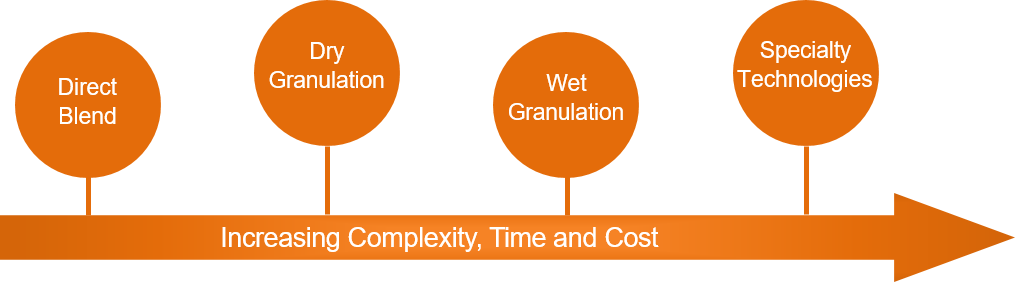
Manufacturing a drug within an accelerated timeline is a significant endeavor, and it is important to make sure that the manufacturing process is robust and compatible during commercial manufacturing. While simplicity and robustness are keys to avoiding delays and setbacks, the manufacturing approach should not be designed solely around speed. It should be evaluated and selected based on the possibility of different dosage forms, low-risk stability, API variability, and applicability of a large range of drug loads.
A complex manufacturing process can result in complicated technical transfers which can exhaust API availability and result in longer development timelines. Therefore, it is advantageous to streamline the initial manufacturing process to maximize the API and decrease the time to commercial manufacturing.
Communication and knowledge transfer are critical to maintaining an accelerated timeline. If possible, co-location of both development and manufacturing is ideal for streamlining processes and ease of communication. Additionally, for accelerated timelines, technical transfers should be done before the registration batch(es) to ensure the team fully understands the process performance qualification (PPQ). It should be noted that registration batches that are manufactured at the site may help avoid and/or isolate future pitfalls and delays.
Regulatory requirements should be kept at the forefront throughout the accelerated program, including the CMC requirements and pre-approval inspection requirements. Regulators will want to see that quality-by-design (QbD) has been transferred to the manufacturing site to estimate the potential impact of any post-approval changes. A thorough end-to-end risk assessment is a useful tool for effective technical transfers because it takes into consideration all development phases- from API selection, formulation development, to packaging and distribution. The process design of experiment and understanding of critical process parameters are also essential for successful manufacturing and is key for PPQ for an accelerated timeline. Consider partnering with a regulatory agency to get their input and build strategies that can assist in streamlining the process. By focusing on both central and district representatives, the sponsors can gain insight on the best practices, effective strategies, and proven past approaches.
OVERCOMING HURDLES IN SCALE-UP AND POST-LAUNCH CONSIDERATIONS
As you would expect, thorough planning is critical to the success of accelerated timelines, especially when it comes to the scale-up processes and post-launch changes.
Consider, for instance, the scenario of increasing production as a label expands post-launch.
In an accelerated timeline, this puts more pressure on manufacturing to expedite the validation process and strategies to select and deliver at the right scale. It is also important to ensure that the process was designed earlier for scalability variations. For example, a semi-continuous or continuous process such as roller compaction, if designed and justified correctly, may result in fewer PPQ batches.
Packaging should also be considered as part of the life cycle planning strategy. Different packaging considerations may be required at higher volumes. Packaging runs increase as the volume increases, so a different, yet appropriate technique could be chosen that changes the entire labeling strategy (i.e., bright stock), which requires packing in unlabeled primary containers. This reduces longer lead times for “typical” bulk shipments under continuous changes. A successful strategy for post-approval changes should also consider expiry dating, which is determined from long-term stability studies, and can affect supply chain considerations and organizational assumptions regarding batch sizes and product mix. Additionally, the volume and packaging changes can have an impact on the serialization process.
For the post-launch commercial life cycle of a product, the development team should consider the possibility of a line extension or adding additional dosage strengths. For this, it is important to leverage prior knowledge of QbD and quality risk assessments to reduce regulatory impact and seamless transition to manufacturing sites. Creating a business continuity plan that streamlines further batch and process considerations can mitigate errors in communication and help post-launch/additional commercial decisions. Reliable supply chain management, the selection of appropriate raw materials, and efficient drug manufacturing and packaging sites are all key components in this next stage of the product life cycle. Additionally, collaborative and clear communication of strategies, guidelines, and goals at all stages of the process can help mitigate risks, setbacks, conflicts, and other potential issues during the development and manufacturing process.
As you would expect, thorough planning is critical to the success of accelerated timelines, especially when it comes to the scale-up processes and post-launch changes.
Consider, for instance, the scenario of increasing production as a label expands post-launch.
In an accelerated timeline, this puts more pressure on manufacturing to expedite the validation process and strategies to select and deliver at the right scale. It is also important to ensure that the process was designed earlier for scalability variations. For example, a semi-continuous or continuous process such as roller compaction, if designed and justified correctly, may result in fewer PPQ batches.
Packaging should also be considered as part of the life cycle planning strategy. Different packaging considerations may be required at higher volumes. Packaging runs increase as the volume increases, so a different, yet appropriate technique could be chosen that changes the entire labeling strategy (i.e., bright stock), which requires packing in unlabeled primary containers. This reduces longer lead times for “typical” bulk shipments under continuous changes. A successful strategy for post-approval changes should also consider expiry dating, which is determined from long-term stability studies, and can affect supply chain considerations and organizational assumptions regarding batch sizes and product mix. Additionally, the volume and packaging changes can have an impact on the serialization process.
For the post-launch commercial life cycle of a product, the development team should consider the possibility of a line extension or adding additional dosage strengths. For this, it is important to leverage prior knowledge of QbD and quality risk assessments to reduce regulatory impact and seamless transition to manufacturing sites. Creating a business continuity plan that streamlines further batch and process considerations can mitigate errors in communication and help post-launch/additional commercial decisions. Reliable supply chain management, the selection of appropriate raw materials, and efficient drug manufacturing and packaging sites are all key components in this next stage of the product life cycle. Additionally, collaborative and clear communication of strategies, guidelines, and goals at all stages of the process can help mitigate risks, setbacks, conflicts, and other potential issues during the development and manufacturing process.
Regardless of the assigned FDA expedited approval designation and approach, the key action items should be: the assurance of a detail-oriented plan; the investigation for the possibility of inefficient processes, miscommunication, or other potential setbacks; and the adaptation of a “forward-thinking” approach that will assist in any future challenges and opportunities for growth with each project.
SUMMARY
Regardless of the assigned FDA expedited approval designation and approach, the key action items should be: the assurance of a detail-oriented plan; the investigation for the possibility of inefficient processes, miscommunication, or other potential setbacks; and the adaptation of a “forward-thinking” approach that will assist in any future challenges and opportunities for growth with each project.





