Overview
Drug developers have to take into account several factors to position a pre-clinical drug candidate successfully for first-in-human clinical trials. This executive summary will outline the pre- formulation studies necessary to chart out the best development path for a drug candidate. Additionally, it will describe clinical formulation options, including oral and injectable routes, for both short-term and long-term success.
Molecule Characterization
Developing a new prescription drug that receives marketing approval is a costly process estimated at $2.6 billion. The significant attrition rate caused by insufficient efficacy and safety is the primary reason for these high costs. Therefore, drug developers must carefully balance the time and resources allocated to various activities such as API synthesis, pre-formulation studies, pharmacokinetics, animal toxicity studies, formulation development, and clinical material supplies. The initial stage in the development of a new molecule is the pre-formulation studies, also known as a “developability assessment.” This stage is critical in determining the molecule’s potential for successful development and entails various activities, such as assessing the molecule’s physicochemical properties, solubility, stability, and compatibility with excipients. To select the most suitable formulation and route of administration for the drug candidate scientists must consider the following factors:
- Solubility at different pH levels and in bio-relevant media
- Dissolution behavior
- Stability of the drug molecule under various conditions, including temperature, light, humidity, andpH
- molecular weight
- crystallinity
- solubility at
- solid state properties: polymorphism, melting point, rheology, bulk density, water sorption behavior
- Octanol-water partition coefficient/Lipophilicity
- acid dissociation constant
- Solid-state properties including salts and polymorphs, melting point, moisture absorption profile, bulk density, and flow properties.
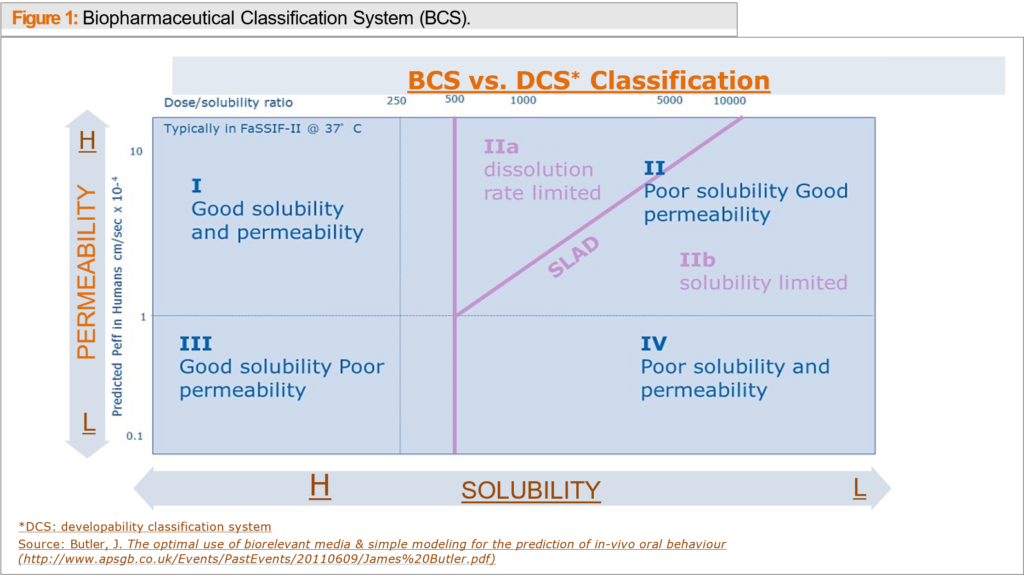
To guide the development of a drug’s formulation, scientists use data to evaluate its oral bioavailability. The FDA’s Biopharmaceutics Classification System (BCS) or Developability Classification System (DCS) are commonly used to categorize molecules into four classes based on their solubility and permeability. (see Figure 1).
Dosing Options for Testing Oral Formulations
Developers working with oral formulations have several Phase I options that can accelerate a drug’s introduction into clinical trials. One approach is Powder-in-Bottle (PIB), which can progress a molecule quickly into the clinic within one to four weeks and at a relatively low cost, with minimal development and GMP manufacturing support needed. Analytical requirements for PIB are minor and clinical dosing is flexible.
Similar to PIB, Powder-in-Capsule (PIC) allows for quick progression of a molecule into the clinic with minimal development and manufacturing support required, helping to save time and cost. Unlike PIB, PIC eliminates taste-related patient compliance issues, and has a finished look resembling a final dosage form. Development timelines are typically shorter with PIC since minimal material requirements and development time is required. However, low, or high strengths may require manufacturing at a CDMO rather than the clinical site. A capsule shell disintegration or dissolution method must also be developed and tested for PIC.
Analytical requirements for a PIC are greater than for a PIB because, in addition to the active pharmaceutical ingredient (API), you must also consider the capsule shell. As a result, you will need to develop and test disintegration and dissolution methods specific to the capsule shell. The capsule shell can affect the release of the API, so it’s essential to consider the shell material, thickness, and other properties. It’s also important to note that the capsule shell can impact the stability of the API, especially if it is sensitive to moisture or other environmental factors.
When it comes to presenting the drug in capsule form for clinical trials, there are a few options to consider. Capsules usually come in sizes 4 to 00 and can be filled at the clinical site as needed or filled manually, semi-automatically, or automatically at a CDMO. It is important to consider the dose range and clinical site resources when choosing a filling method. For doses less than 5 mg, specialty equipment may be necessary, and for doses exceeding 500 mg, multiple capsules may be required.
According to research, over 90% of small-molecule drug candidates are classified as DCS II or IV due to poor solubility. Poorly soluble compounds pose several challenges, including non-linear dose proportionality, variable pharmacokinetic data, and limited toxicity coverage, which can affect human dosing prediction and studies. To overcome these challenges, bio- enhancing formulations are necessary to achieve consistent and good exposure with over 30% bioavailability in various animal species and humans.
Salt and Polymorph Selection
Creating salt forms is an efficient technique to enhance the solubility of molecules with ionizable functional groups. Over 50% of the small-molecule drugs available on the market have been developed as salt forms. By carefully selecting salts and polymorphs, drug developers can improve a compound’s physical properties, such as solubility and stability, as well as facilitate API isolation and purification. These strategies can help to improve the chemical stability of a particular molecule, making it more suitable for use as a pharmaceutical product.
The free acid form of aripiprazolel has limited solubility, which can affect its bioavailability. To address this drugmakers utilized various salt forms and a free acid including aripiprazole lauroxil, aripiprazole hydrochloride, and aripiprazole laurate. These salt forms have improved solubility, lead to more consistent pharmacokinetics and improved patient outcomes.
Another example is ibuprofen which is a non-steroidal anti-inflammatory drug (NSAID) that is used to treat pain, fever, and anti- inflammation. The free acid form of ibuprofen has poor solubility and bioavailability, which limits its effectiveness. However, when ibuprofen is converted into its lysine salt form, it has improved solubility and can be delivered intravenously for more rapid pain relief.
One more example of how drugmakers created salt forms to enhance molecules is the drug lurasidone, which is used to treat schizophrenia and bipolar depression. The free base form of lurasidone has poor solubility in water, which limits its absorption and bioavailability in the body. To improve the drug’s therapeutic profile, the drugmaker developed a salt form of lurasidone by combining the free base with hydrochloric acid to create lurasidone hydrochloride. This salt form has higher solubility in water, which enhances its absorption and bioavailability, improving the drug’s therapeutic profile.
Oral Formulation Strategies:
From Preclinical to Clinical Toxicity Studies quality formulation that provides consistent and maximal exposure while minimizing complexity is essential for good laboratory practice (GLP) toxicity studies. It is also recommended to use the same lot of APIS as planned for first-in-human studies, if the compound is physically representative. If this is not possible, performing an in-use stability study can be helpful. However, the preclinical formulation may differ from the clinical formulation used in first-in-human studies. Toxicology studies conducted in preclinical phases require a higher linear exposure range compared to the dosing limit intended for humans. This is necessary to determine the observed adverse effect level and the maximum tolerated dose of the drug candidate.
Optimizing the solubility and physiochemical stability of a molecule is crucial during the drug development process to ensure a safe and non-toxic formulation that can be used for extended periods of time. Solubilizing agents used may vary during different stages of development, from initial screening to pre-clinical and clinical formulations. It’s essential to establish GLP product manufacturing and analytical support at this stage.
Several factors should be considered as the drug product progresses to Phase 1 human trials, including placebos, taste, equipment, dosing flexibility, corporate value, BCS classification, time, cost, and the compound’s advantages and disadvantages. By Phase 2 trials, the formulation should be near the final dosage form needed for the study population and commercial manufacturing.
When choosing among different oral formulations or an injectable formulation, it’s important to take into account the compound’s BCS classification, physiochemical characteristics, the amount of API available, and the resources required for formulation development.






