
INTRODUCTION
The development of orally administered small molecules is becoming more challenging as an increasing number of molecules in pipelines have poor aqueous solubility and bioavailability. This article will introduce an efficient development process that can help deliver the right clinical candidate, optimal formulation strategy and the most appropriate dosage form to clinic. It will also review some established data-driven approaches to formulation development, focus on the value proposition for discovery biopharmaceutics and discuss a preformulation toolkit for drug discovery.
VALUE PROPOSITION FOR DISCOVERY BIOPHARMACEUTICS
Drug discovery and development is a lengthy and costly undertaking, but well-planned early stage work can help de-risk development, reduce attrition, avoid excessive late phase efforts and decrease delays in patient access. The drug discovery process begins with the validation of drug targets and pathways, then moves into assessment of
pharmacokinetic-pharmacodynamic responses and/or efficacy in preclinical models, establishment of structure activity relationships (SAR), and identification and optimization of lead compounds.
Discovery biopharmaceutics plays different roles at each stage of discovery and development. In the early phases of discovery, biopharmaceutics groups often support compound screening with formulation development for low-dose pharmacokinetic (PK) studies and provide physical chemical assessments with the goal of achieving exposure in preclinical experimental models. In the optimization phase, the biopharmaceutics group provides a full developability assessment and physical form recommendations to ensure consistent exposure is achieved across subsequent studies.

As the program moves forward to toxicology studies, it is essential to develop formulations that can achieve sufficient exposure at high doses to establish the therapeutic index for the development candidate compounds. At each stage of development, different aspects of exposure are addressed, and it is important to understand the factors that can affect exposure.
As an orally dosed compound transits through the gastrointestinal (GI) tract, absorption, gut metabolism and liver metabolism will all influence how much of the compound reaches the bloodstream. The fraction of the oral dose that is absorbed and avoids gut and liver metabolism to reach the systemic circulation is the bioavailable fraction. Formulators can only impact the fraction of the dose absorbed, thus it is important to understand the processes that affect the bioavailability of a compound and where formulation optimization can resolve the issue, or where a new candidate compound needs to be selected.
The case study illustrated in figure 1 shows two compounds, each with a bioavailability of 24%. Compound 1 is 30% absorbed with 80% of the absorbed dose escaping gut and liver metabolism. Compound 2 is 80% absorbed with 30% of the absorbed dose escaping gut and liver metabolism. Since only 30% of compound 1 is absorbed and a large portion of the absorbed dose escapes metabolism, it is possible to improve its bioavailability by investigating the root cause of the low fraction absorbed and selecting a formulation approach that overcomes that issue. Compound 2, on the other hand, already has an 80% fraction absorbed; therefore, further improvements in fraction absorbed are unlikely to significantly improve bioavailability, and as most of the absorbed fraction is metabolized, it is advisable to select a different candidate.
PREFORMULATION TOOLKIT FOR BIOPHARMACEUTICAL RISK ASSESSMENT IN DRUG DISCOVERY
Several simple tools can be used for biopharmaceutical risk assessment early in discovery, which include the developability classification system (DCS) and preclinical dose number (PDo) evaluation. These tools categorize new chemical entities by the key factors that limit oral absorption and have the advantage of requiring minimal data such as in vitro permeability, solubility, and the dose. The solvent-shift solubility assay assesses whether a compound can maintain supersaturation in intestinal fluids even if it has poor thermodynamic solubility. Additionally, absorption modeling (e.g., using GastroPlus®) can help predict whether the compound can achieve the desired exposure in toxicology studies and identify the critical factors and parameters for oral absorption.
- Compounds that fall in DCS class I should achieve sufficient exposure using conventional formulations without solubility or permeability limitations. DCS IIa compounds can achieve sufficient exposure with simple strategies that improve the dissolution rate.
- DCS class III compounds are permeability limited in their absorption and formulation is unlikely to affect exposure.
- DCS class IIb compounds are solubility limited with high permeability. Exposureof compounds in this class can be achieved using an enabled formulation.
- DCS class IV compounds are solubility limited and have low permeability and are therefore the highest risk category for poor oral absorption. These compounds will likely require an enabling technology.
Many drug candidates are weak bases that have good solubility in the low pH of the stomach but have poor solubility at the higher pH conditions of the intestine where most commonly drug absorption occurs. At the higher pH, they can precipitate, leading to poor absorption and inconsistent exposure.
The solvent-shift solubility assay helps prioritize compounds that maintain high supersaturation when switching from the low pH in the stomach to higher pH conditions in the intestines. Compounds that can maintain supersaturation are more likely to have good exposure when dosing in vivo.
Absorption modeling helps formulators understand what parameters are limiting exposure and evaluate the risk of seeing non- proportional dose exposure in PK studies.
In the preclinical stage, it can help validate early in vivo PK models, determine absorption limitation, and aid in toxicology study and formulation selection.
Based on the early PK data and physiochemical characteristics of a given compound, GastroPlus software can be used to perform multi-parameter sensitivity analysis. The software can help scientists understand the effects of solubility, permeability, particle size, or dose on the fraction absorbed. For example, in the development of a suspension formulation for animal studies, it is critical to select an appropriate particle size to ensure optimal exposure of the compound. PBPK modeling, with the inputs of available data, can be used to predict the effect of increasing dose and varying particle size on the expected fraction absorbed and help select a suitable particle size range for the compound in the formulation.
PBPK modeling can also help rank developmental compounds according to the risk of poor exposure from conventional formulations. A low risk compound has good predicted exposure across the toxicology and anticipated human dose range when delivered using a conventional formulation. Medium risk compounds may have poor exposure from a suspension or conventional tablets, but they can maintain supersaturation and modeling shows absorption can be improved.
These compounds have the potential to achieve target exposures using enabling technologies and further formulation development is recommended. High risk compounds will have either high clearance or poor solubility and poor supersaturation potential. They will be very difficult to develop into a formulation that will improve exposure. When high risk compounds are identified, it is recommended to terminate their development and go back to the optimization phase to use SAR to improve upon these intrinsic properties.
A recent case study demonstrates the utility of PBPK modeling in formulation selection for dose ranging toxicology studies. The lead compound showed dose proportional PK in rats and the team wanted to move forward at risk to a two week toxicology study in both rats and dogs. The PBPK model and in vitro solubility data can predict the impact of the difference in biology between rats and dogs on the effective duodenal solubility of the API. In this case, it was important to consider the duodenal bile salt concentrations in the two species, where dogs in the fasted state have a four-fold lower bile salt concentration compared to rats.
Due to the difference in bile salt levels, the effective in vivo solubility in a dog will be about four-fold less than in a rat. In this scenario, dosing the dogs with the API formulated as a simple suspension may lead to solubility limited exposure. Before moving forward with the toxicology study in dogs, it was recommended to first confirm the appropriate formulation approach that can achieve the required exposure.
In the clinical phase, PBPK modeling can be used to help predict first in human (FIH) doses, understand dose exposure relationships, formulation bridging, in vitro/in vivo correlations and predicting absorption in special populations.
STRATEGIES TO ACCELERATE YOUR MOLECULE’S DEVELOPMENT PATH
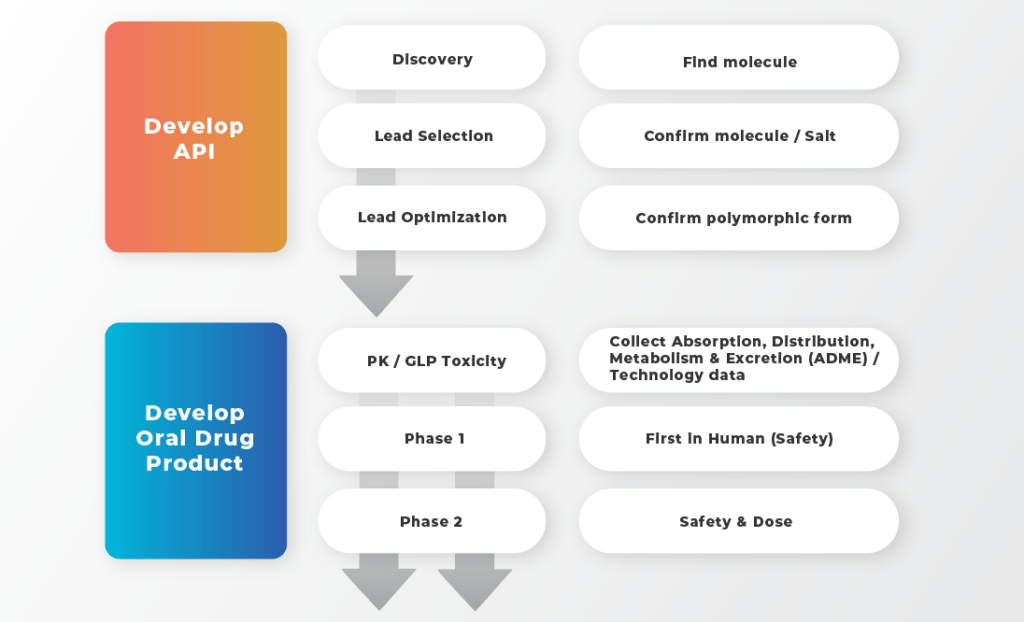
With the focus on small molecules for oral development, strategies capable of accelerating the development path can be broken into three areas: selecting the right molecule, right formulation technology, and the most appropriate dosage form.
The top section in blue illustrates the development of the API. Achieving optimal drug targeting and potency in the controlled laboratory setting of the medicinal chemist is very different than in a dynamic in vivo environment of the patient. It is therefore important that the medicinal chemist engages with the drug product formulator early on.
During the development process, understanding the bioavailability of a drug candidate is critical to predict its success as an oral drug candidate. Bioavailability determination in preclinical studies requires the administration of the drug via both the intravenous (IV) and oral routes and then comparing the resultant PK profiles.
The IV route is used as the comparator for determining oral bioavailability as it inherently has 100% bioavailability since the drug is delivered directly into the blood stream.
When dosing animals in these early studies, it is important to collect reliable data, as this data will help build early PBPK models that will drive future development decisions. Sometimes, mistakes can be made when formulating for early PK studies and toxicology studies:
- Many small molecule drugs in development will fall into the DCS IIb category where their equilibrium solubility severely limits their absorption potential. A study using an oral suspension for a DCS IIb molecule will not provide the exposure required to understand the attributes of the molecule as a drug. Formulation of the candidate using enabling technologies will be needed to improve solubility and ensure adequate exposure in animals.
- During early formulation development, it is important to evaluate the API’s solubility and risk of precipitation in simulated gastric fluid and simulated intestinal fluid. The drug not only needs to be in solution, but also stayin solution as it moves throughout the intestinal tract.
- It is important to understand the properties of the excipients and vehicles.
- used to dose the animals. A perceived easy solution for dosing animals is the administration of the drug dissolved in dimethyl sulfoxide (DMSO). DMSO may be able to solubilize a wide range of molecules, however, it can alter the drug absorption process and may skew the data collected. It is important to keepin mind that not all excipients are truly inactive in the body.
When preparing injectable formulations, it is critical to use well tolerated excipients that can adequately solubilize the API and manage the risk of precipitation in plasma. Precipitation risks are critical with injectables, as there can be serious adverse events if the drug precipitates after injection.
Using optimized preclinical formulations based on our understanding of the DCS classification of the drug will ensure reliable pharmacokinetic data. Subsequent PBPK modeling of these early studies can be used to rank developability potential of lead candidates.
Selecting the right drug formulation technology
As there are no solubility or permeability issues with DCS I molecules, the focus is on a formulation strategy for DCS II molecules. DCS IIa molecules are dissolution rate limited. Use of pharmaceutical salts can help stabilize an API, improve solubility and may be a good choice for moving a DCS IIa to a DCS I.
However, if enabling techniques are still needed, using the salt form can possibly introduce unwanted interactions with equipment or add unwanted bulk to the formulation. Depending on the properties of the salt and the technique needed to improve bioavailability, formulators may prefer to work with a free form of the molecule. Additional approaches for DCS IIa molecules include micronization to increase surface area and utilizing a hydrophilic co-solvent.
A surfactant or polymer can be added to assist in wetting and dispersion of the particles. When utilizing micronization, however, the API must be monitored for possible form change since energy is being introduced into the system to break down the particles. Flow properties may be changed as well.
The hydrophilic co-solvent approach presents the drug in a liquid form which overcomes the need for drug particles to dissolve in the intestines. However, this approach does not contain any components that can help maintain the drug in solution and is not a reliable strategy for DCS IIb molecules.
DCS IIb molecules are equilibrium solubility limited. The two most common DCS IIb approaches are amorphous systems and lipid based systems. Although some molecules can work in either system, the screening process usually identifies the best option. Amorphous systems are good for highly crystalline drugs (“brick dust” molecules). Spray drying and hot melt extrusion are typically used for producing a solid dispersion that can be made into tablets or capsules as the final dosage.
Lipid based systems are well suited for high log P molecules, while also providing additional benefits like protection from oxidation and improved content uniformity, which is especially helpful for low dose drugs. Liquid APIs can be easily incorporated into lipid based systems as well.
To create a solid dispersion using spray drying, the drug and polymer are dissolved in an organic solvent to break down the crystalline lattice of the drug and then the resultant solution is sprayed into a drying chamber to form amorphous solid particles as the solvent evaporates. Mixed with additional excipients, the powder is usually pressed into a tablet.
A main consideration for spray drying is that the drug must be soluble in a solvent. Also, at a commercial scale, large amounts of solvent would be necessary. With hot melt extrusion, instead of dissolving in a solvent, the API and the polymer are melted together, extruded and milled. If molecules are thermally labile, then the high temperatures they are exposed to in processing can be an issue. The benefit of an amorphous solid dispersion is that the polymer surrounding the API helps maintain the state of supersaturation once the dosage form dissolves, preventing recrystallization or possible precipitation.
Lipid based formulations come in many variations, including digestible formulations and self-emulsifying systems, which are often commercialized as soft gel capsules. For digestible lipid formulations, lipid droplets containing the drug are digested by gastric and intestinal fluids in the presence of bile salts and lead to a slower absorption process. For a self-emulsifying drug delivery system (SEDDS), the formulation contains surfactants that produce a nano/microemulsion when diluted in intestinal fluids and can have rapid absorption as compared to digestible lipid formulations.
The suitability of a digestible formulation can be assessed by evaluating the API’s solubility in digestible lipid products versus standard biorelevant media. A lipid approach may impact how the body will take up the drug, its gut wall permeability, and even promote lymphatic uptake. Therefore, it is recommended to use in vivo studies to fully evaluate the potential of the formulation approach.
API sparing techniques. When moving from candidate selection to preformulation/ formulation and in vivo studies, larger amounts of the API will be needed. Therefore, the more work that is done in earlier phases, the more efficiently a limited API supply can be utilized. It can begin by front loading at the preformulation and formulation selection stages to decrease reliance on in vivo data. Hycon’s vitro technology screening tools can help develop optimal formulation with minimal API consumption, as illustrated below.
Before taking that final step of in vivo studies, prototypes can be characterized in rank order to ensure the best candidates progress. One tool important to these studies is the “Micro” Dissolution Tool, which utilizes small sample sizes, making it an ideal tool where API supply is limited. It provides the ability to analyze multiple samples with very small amounts of material using in-situ detection.
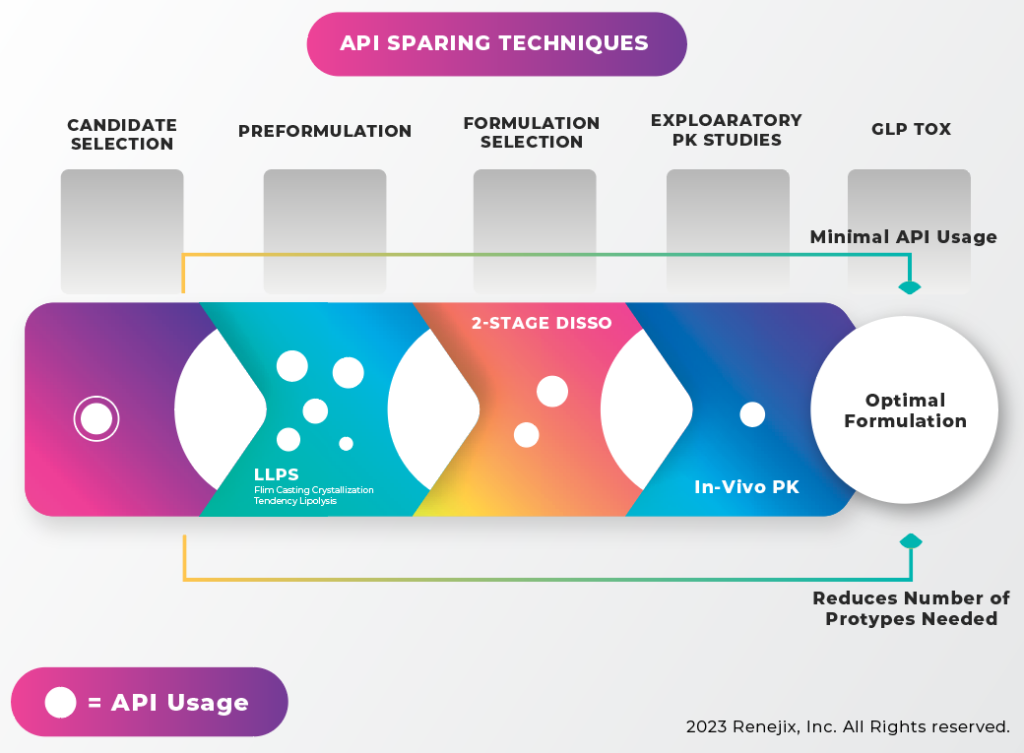
As a case study, let’s examine the use of liquid-liquid phase separation (LLPS) experiments and micro-dissolution to guide formulation development and prototype selection.
The LLPS assay was used to screen several polymers and determine their impact on supersaturation of an API. The results showed phase separation just before precipitation would occur. The LLPS data showed the expected supersaturation concentration of the API to be just under 200 mg/mL. The micro-dissolution was conducted over an extended time to help discriminate between the polymers investigated. The micro-dissolution results showed that although the HPMCE3 polymer could achieve and maintain supersaturation for some time, the API eventually began to precipitate, so the polymer was ruled out for use in future studies (FIGURE 5). Another way to adapt the micro-dissolution apparatus is to look at flux, or the movement of the drug across a membrane. Using a two-stage dynamic media conversion of simulated gastric fluid to fasted state simulating intestinal fluid with pancreatic lipase, the ability of the drug to disperse from a formulation, stay dissolved, and cross a simulated membrane, can be examined. In vitro-in vivo correlation experiments have shown that MicroFLUX data correlates with in vivo data. Although it may not be able to predict in vivo exposure, MicroFLUX experiments can be used to rank the prototypes and select those that are most likely to succeed in vivo.

Selecting the right dosage form
selecting the right dosage form by pairing the formulation technology with the right dosage form design. Powder-in-a-bottle (PiB) can be a very simple, flexible approach that can be dosed as a solution or suspension depending on attributes of the drug and diluent.
Drug- in-a-capsule is also very simple if the drug is soluble. For DCS II compounds, a formulated capsule or tablet, with the assistance of excipients to help improve the performance of the API, is preferred. A summary of PiB, drug-in-capsule (PiC or DiC), and formulated capsules or tablets as possible oral dosing options is outlined in FIGURE 6.
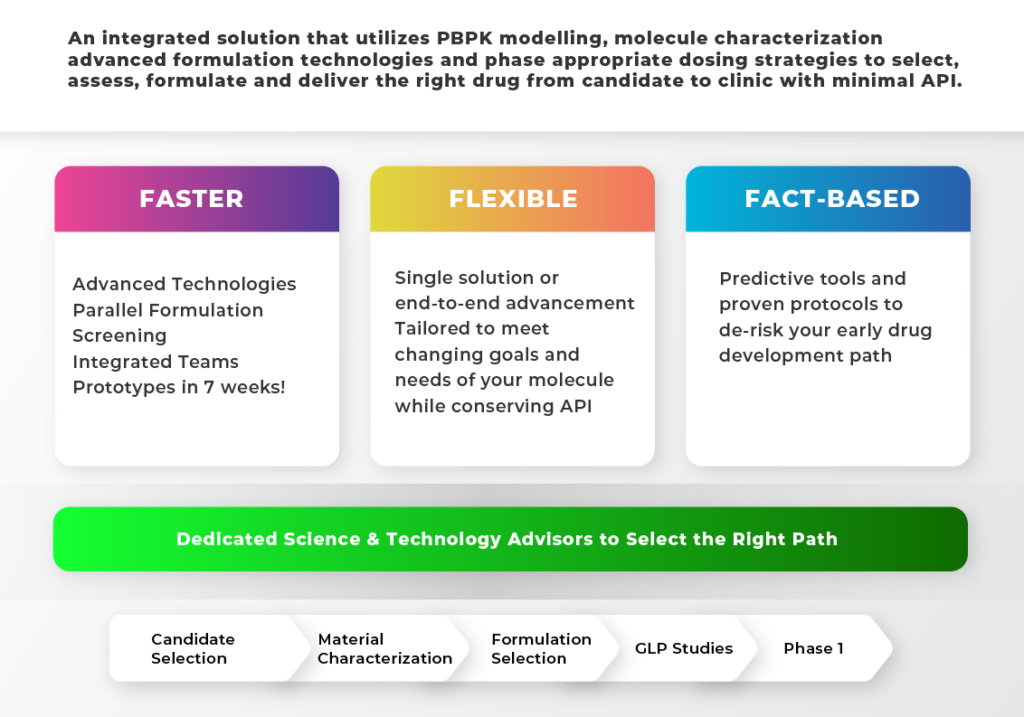
We use a science-based approach to identify the right drug molecule, then pairs it with the right formulation technology and dosage form to ensure accelerated path to clinic, with optimal clinical outcomes for patients..
Drug discovery and development can be a lengthy, costly and risky undertaking. Predictive tools such as the DCS and PBPK modeling and API-sparing in vitro techniques can help accelerate the delivery of therapeutics to clinic. The DCS can help formulators assess what technology to apply to ensure that the bioavailability of the drug will be optimally enhanced. PBPK analysis examines how the drug performs in early animal models, and helps scientists understand the main drivers of bioavailability. API-sparing in vitro formulation characterization tools can help select the optimal formulation technology while conserving limited supplies of API. On their own, these techniques generate useful data, however, evaluating data from all these tools provides the most benefit. Hycon’s scientists not only have the tools and expertise to generate and interpret such data, but they also offer parallel formulation technology screening and a successful track record of optimizing thousands of molecules using wide variety of formulation technologies and dose forms to help de-risk and accelerate your early development programs.
Conclusion
Drug discovery and development can be a lengthy, costly and risky undertaking. Predictive tools such as the DCS and PBPK modeling and API-sparing in vitro techniques can help accelerate the delivery of therapeutics to clinic. The DCS can help formulators assess what technology to apply to ensure that the bioavailability of the drug will be optimally enhanced. PBPK analysis examines how the drug performs in early animal models, and helps scientists understand the main drivers of bioavailability. API-sparing in vitro formulation characterization tools can help select the optimal formulation technology while conserving limited supplies of API. On their own, these techniques generate useful data, however, evaluating data from all these tools provides the most benefit. Hycon’s scientists not only have the tools and expertise to generate and interpret such data, but they also offer parallel formulation technology screening and a successful track record of optimizing thousands of molecules using wide variety of formulation technologies and dose forms to help de-risk and accelerate your early development programs.





